
Recombinación homóloga

La recombinación homóloga es un tipo de recombinación genética en la que las secuencias de nucleótidos se intercambian entre dos moléculas similares o idénticas de ADN. Es la más utilizada por las células para reparar roturas nocivas que se producen en ambas hebras de ADN, conocidas como rupturas de doble hebra. La recombinación homóloga también produce nuevas combinaciones de secuencias de ADN durante la meiosis, el proceso por el cual las células eucariotas hacen gametos, como espermatozoides y óvulos en los animales. Estas nuevas combinaciones de ADN representan la variación genética en la descendencia, que a su vez permite que las poblaciones se adapten durante el curso de la evolución.[1] La recombinación homóloga se utiliza también en la transferencia horizontal de genes para el intercambio de material genético entre diferentes cepas y especies de bacterias y virus.
A pesar de que la recombinación homóloga varía ampliamente entre los diferentes organismos y tipos de células, la mayoría de las formas involucran las mismas etapas básicas. Después de que se produce una ruptura en la doble cadena, las secciones de ADN en torno a los extremos 5' de la ruptura se cortan en un proceso llamado resección. En la invasión de hebra, el paso que sigue, es que un extremo saliente 3' de la molécula de ADN rota "invade" una molécula de ADN similar o idéntica que no está rota. Después de la invasión de la hebra, la secuencia adicional de acontecimientos puede seguir cualquiera de las dos vías principales que se mencionan a continuación (ver Modelos); la vía DSBR (reparación de roturas de doble cadena) o la vía SDSA (síntesis de línea de recocidos dependientes). La recombinación homóloga que se produce durante la reparación del ADN tiende a dar lugar a productos que no son de cruce, en decir, la restauración de la molécula de ADN dañada, tal como existía antes de la ruptura de la doble cadena.
La recombinación homóloga se conserva a través de los tres dominios de la vida, así como los virus, lo que sugiere que se trata de un mecanismo biológico casi universal. El descubrimiento de genes para la recombinación homóloga en protistas —un grupo diverso de microorganismos eucariontes— ha sido interpretado como evidencia de que la meiosis surgió de manera temprana en la evolución de los eucariontes. Ya que su disfunción ha sido fuertemente asociada con una mayor susceptibilidad a varios tipos de cáncer, las proteínas que facilitan la recombinación homóloga son temas de investigación activa. La recombinación homóloga se utiliza también en "gene targeting", una técnica para la introducción de cambios genéticos en organismos objetivo. Para su desarrollo de esta técnica, Mario Capecchi, Martin Evans y Oliver Smithies fueron galardonados con el Premio Nobel de Fisiología o Medicina en el 2007; Capecchi[2] y Smithies[3] descubrieron de forma independiente las aplicaciones de las células madre embrionarias de ratón, sin embargo, los mecanismos altamente conservados se basa el modelo de reparación de DSB, incluyendo la integración homóloga uniforme de ADN transformado (terapia génica) que fue mostrado por primera vez en experimentos de plásmidos por Orr-Weaver, Szostack y Rothstein.[4][5][6] La investigación de la OSD plásmido inducido, utilizando γ-irradiación[7] en los años 1970's-1980, condujo a experimentos posteriores utilizando endonucleasas (por ejemplo, I-Scel) que cortan los cromosomas para la ingeniería genética de células de mamíferos, donde la recombinación no homóloga es más frecuente que en levadura.[8]
Historia y descubrimiento[editar]

A principios de 1900, William Bateson y Reginald Punnett encontraron una excepción a uno de los principios de la herencia descritos originalmente por Gregor Mendel en la década de 1860. En contraste con la noción de Mendel que los rasgos son surtidos de forma independiente cuando pasan de padres a niños- por ejemplo para que el color del pelo de un gato y su longitud de la cola son heredados independientes entre sí- Bateson y Punnett mostraron que ciertos genes asociados con rasgos físicos pueden ser heredados juntos o genéticamente vinculados.[9][10] En 1911, después de observar que los rasgos ligados en ocasiones podrían ser heredado por separado, Thomas Hunt Morgan sugirió que los "cruces" pueden ocurrir entre los genes ligados,[11] donde uno de los genes ligados se cruza físicamente a un cromosoma diferente. Dos décadas después, Barbara McClintock y Harriet Creighton demostraron que se produce un entrecruzamiento cromosómico durante la meiosis,[12][13] el proceso de la división celular por el cual se hacen espermatozoides y óvulos. En el mismo año que el descubrimiento de McClintock, Curt Stern, mostró que el cruce más tarde llamado "recombinación" se podría también producir en las células somáticas como los glóbulos blancos y células de la piel que se dividen a través de la mitosis.[12][14]
En 1947, el microbiólogo Joshua Lederberg demostró que las bacterias que habían sido asumidas que solo se podrían reproducir asexualmente a través de la fisión binaria, son capaces de recombinación genética, lo que es más similar a la reproducción sexual. En este trabajo se estableció a la E. coli como organismo modelo en la genética,[15] y ayudó a Lederberg ganó el Premio Nobel 1958 en Fisiología o Medicina.[16] Sobre la base de estudios en los hongos, en 1964 Robin Holliday propuso un modelo para la recombinación en la meiosis, que introdujo los detalles clave de cómo el proceso puede funcionar, incluido el intercambio de material entre los cromosomas a través de uniones Holliday.[17] En 1983, Jack Szostak y sus colegas presentaron un modelo que ahora se conoce como la vía DSBR, que representó observaciones no explicadas por el modelo de Holliday.[17][18] Durante la siguiente década, los experimentos en Drosophila, levadura en ciernes y células de mamíferos dieron lugar a la aparición de otros modelos de la recombinación homóloga, llamadas vías SDSA, que no siempre se basan en uniones Holliday.[17]
En eucariontes[editar]
La recombinación homóloga (RH) es esencial para la división celular en eucariotas como plantas, animales, hongos y protistas. En las células que se dividen a través de la mitosis, la recombinación homóloga repara las rupturas de la doble cadena en el ADN causadas por la radiación ionizante o productos químicos que dañan el ADN.[19] Si no se reparan, estas rupturas de doble cadena pueden causar una reorganización a gran escala de los cromosomas en las células somáticas,[20] que se puede convertir en cáncer.[21]
Además de la reparación del ADN, la recombinación homóloga también ayuda a producir diversidad genética cuando las células se dividen en la meiosis para convertirse en gametos: huevo o esperma en animales, polen u óvulos en las plantas, y las esporas en hongos. Lo hace mediante la facilitación del entrecruzamiento cromosómico, en el que las regiones de ADN similares pero no idénticas se intercambian entre cromosomas homólogos.[22][23] Esto crea nuevas combinaciones, posiblemente beneficiosas de genes, que pueden dar a la descendencia una ventaja evolutiva.[24] El entrecruzamiento cromosómico comienza cuando una proteína llamada Spo11 hace una ruptura en la doble cadena específica en el ADN.[25] Estos sitios están ubicados de forma no aleatoria en los cromosomas; por lo general en regiones promotoras intergénicas y preferentemente en dominios ricos en GC.[26] Estos sitios de doble cadena suelen ocurrir en sitios de recombinación, las regiones en los cromosomas que son alrededor de 1.000-2.000 pares de bases de longitud y tienen altas tasas de recombinación. La ausencia de un punto de acceso de recombinación entre dos genes en el mismo cromosoma a significa que esos genes se van a heredar por las futuras generaciones en la misma proporción. Esto representa la vinculación entre los dos genes mayor de lo que sería de esperar, a partir de genes que se clasifican independientemente durante la meiosis.[27]
Momento en el ciclo celular mitótico[editar]

Las rupturas de la doble cadena se pueden reparar mediante recombinación homóloga o por medio de extremos no homólogos (NHEJ). NHEJ es un mecanismo de reparación del ADN que, a diferencia de la recombinación homóloga, no requiere una secuencia homóloga larga para guiar la reparación. Ya sea recombinación homóloga o NHEJ se utilizan para reparar roturas de doble cadena y se determina en gran medida por la fase del ciclo celular. La recombinación homóloga repara el ADN antes de que la célula entra en mitosis (fase M). Se produce durante y poco después de la replicación del ADN, en las fases S y G2 del ciclo celular, cuando las cromátidas hermanas son fácilmente disponibles.[28] En comparación con los cromosomas homólogos, que son similares a otro cromosoma, pero a menudo tienen diferentes alelos, las cromátidas hermanas son una plantilla ideal para la recombinación homóloga, ya que son una copia idéntica de un cromosoma dado. En contraste con la recombinación homóloga, NHEJ es predominante en la fase G1 del ciclo celular, cuando la célula está creciendo, pero aún no está lista para dividirse. Se produce con menos frecuencia después de la fase G1, pero mantiene al menos alguna actividad a lo largo del ciclo celular. Los mecanismos que regulan la recombinación homóloga y NHEJ a lo largo del ciclo celular varían ampliamente entre especies.[29]
Las cinasas dependientes de ciclina (CDKs), que modifican la actividad de otras proteínas mediante la adición de grupos fosfato a (es decir, fosforilación) ellos, son importantes reguladores de la recombinación homóloga en eucariotas.[29] Cuando la replicación del ADN comienza en la levadura, la cinasa dependiente de la ciclina Cdc28 comienza la recombinación homóloga mediante la fosforilación de la proteína Sae2.[30] Después de haber sido activada mediante la adición de un fosfato, la Sae2 utiliza su actividad de endonucleasa para hacer un corte limpio cerca de una ruptura en la doble cadena en el ADN. Esto permite que una proteína de tres partes conocida como el complejo MRX se una al ADN, y comienza una serie de reacciones de proteínas impulsadas que intercambian el material entre dos moléculas de ADN.[31]
Modelos[editar]
Dos modelos principales para la forma en que la recombinación homóloga repara las rupturas de doble cadena en el ADN son la vía de reparación de la ruptura de cadena doble (DSBR) (a veces llamado el modelo de doble cruce Holliday) y la vía de síntesis que depende de la línea del recocido (SDSA).[32]
Los dos caminos son similares en sus primeros pasos. Después se produce una ruptura en la doble cadena, el complejo MRX (complejo MRN en los seres humanos) se une al ADN a ambos lados de la ruptura. A continuación una resección, en la cual el ADN alrededor de los extremos 5' de la ruptura se recorta, y se lleva a cabo en dos etapas distintas. En el primer paso de la resección, el complejo MRX recluta la proteína Sae2. Las dos proteínas a continuación recortan los extremos 5' a ambos lados de la ruptura para crear lados cortos salientes 3' de ADN de una sola hebra. En el segundo paso, 5 '→ 3' de la resección se continúa por la helicasa Sgs1 y las nucleasas Exo1 y Dna2. Una helicasa, Sgs1 "abre" el ADN de doble cadena, mientras que la actividad nucleasa Exo 1 y de Dna2 les permite cortar el ADN de una sola hebra producida por Sgs1.[30]

La proteína RPA, que tiene alta afinidad por ADN de una sola cadena, se une a salientes 3'.[33] Con la ayuda de otras proteínas que median en el proceso, la proteína Rad51 (y Dmc1, en la meiosis) forma entonces un filamento de ácido nucleico y la proteína en la cadena simple de ADN recubierto con RPA. Este filamento de nucleoproteína comienza la búsqueda de secuencias de DNA similares a la de la saliente 3'. Después de encontrar una secuencia de este tipo, el filamento de una sola cadena se mueve en la nucleoproteína (invade) el dúplex ADN receptor similar o idéntico en un proceso llamado invasión de cadena. En células que se dividen a través de la mitosis, el dúplex de ADN receptor es generalmente una cromátida hermana, que es idéntica a la molécula de ADN dañada y proporciona una plantilla para su reparación. En la meiosis, sin embargo, el ADN receptor tiende a ser de un cromosoma homólogo similar no necesariamente idéntico.[32]
Un bucle de desplazamiento (D-loop) se forma durante la invasión de cadena entre el invasor 'hebra 3 y el cromosoma homólogo. Después de la invasión de la hebra, una ADN polimerasa extiende el extremo invasor de la hebra 3' mediante la síntesis de un nuevo ADN. Esto cambia el D-loop a una estructura en forma de cruz conocida como una unión de Holliday. Después de esto, más síntesis de ADN se produce en la cadena invasora (es decir, uno de los salientes 3 'originales), restaurando eficazmente la hebra en el cromosoma homólogo que se desplazó durante la invasión de hebra.[32]
Vía DSBR[editar]
Después de las etapas de la resección, la invasión de cadena y la síntesis de ADN, las vías DSBR y SDSA se vuelven diferentes.[32] La vía DSBR es única ya que en el segundo saliente 3' (que no participó en la invasión de hebra) también forma una unión de Holliday con el cromosoma homólogo. Los dobles uniones Holliday se convierten entonces en productos de recombinación por endonucleasas recortadoras, un tipo de endonucleasas de restricción que corta solo una cadena de ADN. La vía DSBR comúnmente resulta en entrecruzamiento, aunque a veces puede dar lugar a productos que no son de entrecruzamiento; la capacidad de una molécula rota de ADN para recoger las secuencias del donante separado loci se muestra en la levadura mitótica usando plásmidos o inducción de endonucleasa de eventos cromosómicos.[34][35]
Debido a esta tendencia de entrecruzamiento cromosómico, la vía DSBR es un modelo de cómo el entrecruzamiento de recombinación homóloga se produce durante la meiosis.[22]
Ya sea que la recombinación en la vía DSBR resulta en un entrecruzamiento cromosómico, está determinada por la forma en que se corta la doble unión Holliday, o "resuelta". El entrecruzamiento cromosómico se producirá si una unión Holliday se corta en la hebra de cruce y la otra unión de Holliday se corta en la hebra sin cruce (en la figura 4, a lo largo de las puntas de flecha púrpura horizontales están en una unión Holliday y a lo largo de las puntas de flecha naranja verticales en la otra). Alternativamente, si las dos uniones Holliday se cortan en las hebras de cruce (a lo largo de las puntas de flecha púrpura horizontales en ambas uniones Holliday en la Figura 4), a continuación, los cromosomas sin entrecruzamiento serán producidos.[36]
Vía SDSA[editar]
La recombinación homóloga a través de la vía de SDSA se produce en las células que se dividen a través de la mitosis y la meiosis y se traducen en productos sin entrecruzamiento. En este modelo, la hebra invasora 3' se extiende a lo largo del dúplex de ADN por una ADN polimerasa, y se libera en forma de la unión Holliday entre las moléculas donantes y de las ADN receptoras en un proceso llamado migración de la ramificación. La nueva síntesis de la terminación 3' de la hebra invasora es entonces capaz de hibridar con el otro saliente 3' en el cromosoma dañado a través de un apareamiento de bases complementarias. Después del recocido de hebras, a veces puede permanecer una pequeña hoja de ADN. Cualquiera de estas hojas son eliminadas, y la vía SDSA termina con el resellado, también conocida como ligadura, de cualquier hueco de las hebras restantes.[37]
Durante la mitosis, la principal vía de recombinación homóloga para la reparación de las rupturas del ADN de doble cadena parece ser la vía SDSA (en lugar de la vía DSBR).[38] La vía SDSA produce recombinantes no entrecruzados (Figura 4). Durante la meiosis, los recombinantes no entrecruzados también se producen con frecuencia y éstos parecen surgir principalmente también por la vía de SDSA.[38][39] Eventos de recombinación no entrecruzados que se producen durante la meiosis probablemente reflejan casos de reparación de daños de la doble cadena de ADN o de otros tipos de daños de ADN.
Vía SSA[editar]

La vía de una sola hebra recocida (SSA) de recombinación homóloga repara las roturas de las dobles cadenas entre dos secuencias repetidas. La vía de SSA es única, en el sentido de que no requiere una molécula similar o idéntica separada de ADN, como las vías DSBR o SDSA de recombinación homóloga. En cambio, la vía de la SSA solo requiere un único dúplex de ADN, y utiliza las secuencias repetidas como las secuencias idénticas que la recombinación homóloga necesita para su reparación. La vía es relativamente simple en concepto: después de que dos hebras del mismo dúplex de ADN se cortan alrededor del sitio de la rotura de la doble cadena, las dos salientes 3' resultantes se alinean y se recocen entre sí, restaurando el ADN como un dúplex continuo.[37][40]
Como el ADN en torno a la ruptura de la doble cadena se corta de nuevo, las salientes 3' de cadena simple que se producen se revisten con la proteína RPA, que impide que las 3' salientes se peguen a sí mismas.[41] Una proteína llamada Rad52 se une entonces a cada una de las secuencias repetidas a ambos lados de la ruptura, y los alinea para permitir que se fortalezcan las dos secuencias repetidas complementarias.[41] Después del que el recocido se complete, restos de hojas no homólogas de los salientes 3' se cortan por un conjunto de nucleasas, conocidas como Rad1/Rad10, que son llevadas a las hojas de las proteínas Saw1 y Slx4.[41][42] La síntesis de nuevo ADN llena los espacios vacíos, y la ligadura restaura el dúplex de ADN como dos hebras continuas.[43] La secuencia de ADN entre las repeticiones siempre se pierde, ya que es una de las dos repeticiones. La vía de SSA se considera mutagénica ya que resulta en eliminación de material genético.[37]
Vía BIR[editar]
Durante la replicación del ADN, las rupturas de la doble cadena se pueden encontrar a veces en las horquillas de replicación cuando la ADN helicasa abre la cadena molde. Estos defectos se reparan en la vía de la replicación de ruptura inducida (BIR) de la recombinación homóloga. Los mecanismos moleculares precisos de la vía de BIR siguen sin estar claros. Tres mecanismos propuestos tienen a la invasión de cadena como paso inicial, pero difieren en la forma en que el modelo de migración de la D-loop y en fases posteriores de recombinación.[44]
La vía BIR también puede ayudar a mantener la longitud de los telómeros (regiones de ADN en el extremo de los cromosomas eucariontes) en ausencia de (o en cooperación con) la telomerasa. Sin copias de la enzima telomerasa, los telómeros típicamente se acortan con cada ciclo de la mitosis, lo que finalmente bloquea la división celular y conduce a la senescencia. En células de levadura, donde la telomerasa se ha inactivado a través de mutaciones, se han observado dos tipos de células "supervivientes" para evitar la senescencia más de lo esperado por el alargamiento de sus telómeros a través de vías BIR.[44]
El mantenimiento de la longitud del telómero es crítica para la inmortalización celular, una característica clave del cáncer. La mayoría de los cánceres mantienen los telómeros mediante la regulación positiva de la telomerasa. Sin embargo, en varios tipos de cáncer humano, una vía BIR ayuda a sostener algunos tumores, actuando como un mecanismo alternativo de mantenimiento de los telómeros.[45] Este hecho ha llevado a los científicos a investigar si tales mecanismos basados en la recombinación de mantenimiento de los telómeros para impedir fármacos contra el cáncer, como inhibidores de la telomerasa.[46]
En bacterias[editar]

La recombinación homóloga es un proceso importante de reparación del ADN en bacterias. También es importante para la producción de la diversidad genética en las poblaciones de bacterias, aunque el proceso difiere sustancialmente de la recombinación meiótica, lo que daña las reparaciones de ADN y produce la diversidad en los genomas eucariotas. La recombinación homóloga ha sido más estudiada y se entiende mejor por Escherichia coli.[48] Rupturas de doble cadena de ADN en bacterias son reparados por la vía RecBCD de la recombinación homóloga. Rupturas que se producen en solo una de las dos cadenas de ADN, conocidos como huecos de una sola hebra, se cree que son reparados por la vía RecF.[49]
Tanto las vías RecBCD y RecF incluyen una serie de reacciones conocidas como migración de la ramificación, en la que cadenas simples de ADN se intercambian entre dos moléculas entrecruzadas de dúplex de ADN, y de la resolución, en la que se cortan aparte esas dos moléculas entrecruzadas de ADN y se restauran a su estado de doble cadena normal.
Vía RecBCD[editar]
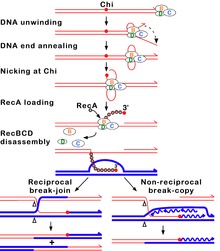
La vía RecBCD es la vía principal de recombinación que se utiliza en muchas bacterias para reparar rupturas en la doble cadena de ADN y las proteínas se encuentran en una amplia gama de bacterias.[51][52][53] Estas rupturas de doble cadena pueden ser causadas por la luz ultravioleta y otras radiaciones, así como mutágenos químicos. Las rupturas de doble cadena también pueden surgir por la replicación de ADN a través de un "nick" o hueco de una solo cadena. Tal situación provoca lo que se conoce como horquilla de replicación colapsado y se repara por varias vías de recombinación homóloga incluyendo la vía RecBCD.[54]
En esta vía, un complejo enzimático de tres subunidades llamadas RecBCD inicia la recombinación mediante la unión a un extremo terminal o casi terminal de una rotura en el ADN de doble cadena. Después RecBCD se une al final del ADN, las subunidades RecB y RecD comienzan descomprimir el dúplex de ADN a través de la actividad de la helicasa. La subunidad RecB también tiene un dominio nucleasa, que corta la única cadena de ADN que emerge del proceso de descompresión. Esta descompresión continúa hasta que RecBCD encuentra una secuencia de nucleótidos específica (5'-GCTGGTGG-3'), conocido como un sitio de Chi.[53]
Al encontrar un sitio Chi, la actividad de la enzima RecBCD cambia drásticamente.[52][55][56] El desenrollo del ADN pausa durante unos segundos y luego se reanuda en más o menos la mitad de la velocidad inicial. Esto es probablemente debido a que la helicasa RecB más lenta, desenrolla el ADN después de Chi, en lugar de la helicasa más rápida RecD, que desenrolla el ADN antes de Chi.[57][58] El reconocimiento del sitio Chi también cambia a la enzima RecBCD de modo que corta la cadena de ADN con Chi y comienza a cargar varias proteínas RecA en el ADN de una sola cadena con el extremo recién generado 3'. El filamento de la nucleoproteína RecA recubierta resultante busca secuencias similares de ADN en un cromosoma homólogo. El proceso de búsqueda induce el estiramiento del dúplex de ADN, que mejora el reconocimiento de homología (un mecanismo denominado como corrección de pruebas conformacional[59][60][61]). Al encontrar tal secuencia, el filamento de nucleoproteína se mueve en el receptor homólogo del dúplex de ADN en un proceso llamado invasión de hebra.[62] El saliente invasor 3' hace que una de las cadenas del dúplex de ADN receptor se desplace, para formar un D-loop. Si se corta el D-loop, otro intercambio de hebras forma una estructura transversal llamada unión Holliday.[53] La resolución de la unión Holliday por alguna combinación de RuvABC o RecG puede producir dos moléculas de ADN recombinante con tipos genéticos recíprocos, si las dos moléculas de ADN que interactúan difieren genéticamente. Alternativamente, el extremo 3' invasor cerca de Chi puede cebar la síntesis de ADN y formar una horquilla de replicación. Este tipo de resolución produce solo un tipo de recombinante (no recíproco).
Vía RecF[editar]
Las bacterias parecen utilizar la vía RecF de la recombinación homóloga para reparar los huecos de cadena simple en el ADN. Cuando la vía RecBCD es inactivada por mutaciones y mutaciones adicionales inactivan las nucleasas SbcCD y Exol, la vía RecF también puede reparar las rupturas del ADN de doble cadena.[63] En la vía RecF la RecQ helicasa desenrolla el ADN y la nucleasa RecJ degrada la hebra con un extremo 5', dejando la hebra con el extremo 3' intacto. La proteína RecA se une a esta hebra y es ayudado tanto por las proteínas RecF, Reco, y RECR o estabilizado por ellas. El filamento de nucleoproteína RecA busca entonces un ADN homólogo e intercambia lugares con la hebra idéntica o casi idéntica en el ADN homólogo.
Aunque las proteínas y los mecanismos específicos implicados en sus fases iniciales son diferentes, las dos vías son similares ya que ambas requieren ADN de cadena simple con un extremo 3' y la proteína RecA para la invasión de hebra. Las vías también son similares en sus fases de migración de la ramificación, en el que se desliza el cruce Holliday en una dirección, y la resolución, en la que las uniones Holliday se escinden separadas por enzimas.[64][65]
El tipo de resolución alternativa, no recíproca también se puede producir por cualquiera de estas vías.
Migración de la ramificación[editar]
Inmediatamente después la invasión de cadena, las de uniones de Holliday se mueven a lo largo del ADN ligado durante el proceso de migración de la ramificación. Es en este movimiento de la unión Holliday en que las pares de bases entre los dos dúplex homólogos de ADN se intercambian. Para catalizar la migración de la ramificación, la proteína RuvA primero reconoce y se une a la unión Holliday y recluta a la proteína RuvB para formar el complejo RuvAB. Dos juegos de la proteína RuvB, que cada uno forma una ATPasa en forma de anillo, se cargan en los lados opuestos de la unión Holliday, donde actúan como bombas gemelas que proporcionan la fuerza para la migración de la ramificación. Entre los dos anillos de RuvB, dos conjuntos de la proteína RuvA se reúnen en el centro de la unión Holliday tal que el ADN en la unión se intercala entre cada conjunto de RuvA. Las hebras de ADN dúplex tanto las de los "receptores" como las de los "donante" se desenrollan en la superficie de RuvA a medida que son guiados por la proteína de un dúplex a otro.[66][67]
Resolución[editar]
En la fase de resolución de la recombinación, las uniones Holliday formadas por el proceso de invasión de hebra se cortan, restaurando así dos moléculas de ADN separadas. Esta escisión se realiza por el complejo RuvAB que interactúa con RuvC, que en conjunto forman el complejo RuvABC. RuvC es una endonucleasa que corta la secuencia degenerada 5'-(A/T)TT(G/C)-3'. La secuencia se encuentra con frecuencia en el ADN, aproximadamente una vez cada 64 nucleótidos.[67] Antes de cortar, es probable que RuvC tenga acceso a la unión de Holliday por el desplazamiento de uno de las dos tetrámeros de Ruva que cubren el ADN.[66] Los resultados de la recombinación en cualquiera de los dos productos "de empalme o "de parche", dependiendo de cómo RuvC escinde la unión Holliday.[67] Los productos de empalme son productos de entrecruzamiento, en el que existe una reordenación del material genético alrededor del sitio de recombinación. Los productos de parche, por otra parte, son productos no cruzados en el que no hay tal reordenamiento y solo hay un "parche" de ADN híbrido en el producto de recombinación.[68]
Facilitando la transferencia genética[editar]
La recombinación homóloga es un método importante para la integración de ADN del donante en el genoma de un organismo receptor en la transferencia genética horizontal, proceso por el cual un organismo incorpora ADN extraño de otro organismo sin ser la descendencia de ese organismo. La recombinación homóloga requiere que el ADN entrante sea muy similar al del genoma receptor, y por lo tanto la transferencia genética horizontal se limita generalmente a bacterias semejantes.[69] Los estudios realizados en varias especies de bacterias han establecido que hay una disminución logarítmica lineal de la frecuencia de recombinación con la diferencia incrementada en la secuencia entre el huésped y el ADN receptor.[70][71][72]
En la conjugación bacteriana, donde se transfiere el ADN entre las bacterias por contacto directo de célula a célula, la recombinación homóloga ayuda a integrar el ADN extraño en el genoma huésped a través de la vía RecBCD. La enzima RecBCD promueve la recombinación de ADN después de que se convierte de una sola cadena de ADN que entra inicialmente a la bacteria a una doble cadena de ADN durante la replicación. La vía RecBCD es también esencial para la fase final de la transducción, un tipo de transferencia horizontal genética en el que el ADN es transferido desde una bacteria a otra por un virus. El ADN bacteriano extraño, es a veces mal incorporado en la cabeza de la cápside de virus bacteriófagos durante el empaquetado del AND vírico de los nuevos bacteriófagos durante la replicación viral. Cuando estos nuevos bacteriófagos infectan otras bacterias, el ADN de la bacteria huésped anterior se inyecta en el cromosoma del nuevo huésped bacteriano como ADN de doble cadena. La enzima RecBCD entonces incorpora este ADN de doble cadena en el genoma de la nueva bacteria huésped.[53]
Transformación bacteriana[editar]
La transformación bacteriana natural implica la transferencia de ADN de una bacteria donante a un bacteria receptora, donde el donante y el receptor son normalmente de la misma especie. La transformación, a diferencia de la conjugación bacteriana y la transducción, depende de numerosos productos de los genes bacterianos que interactúan específicamente para realizar este proceso.[73] Por lo tanto la transformación es claramente una adaptación de las bacterias para la transferencia de ADN. Para que una bacteria se una, tome e integre el ADN del donante en su cromosoma mediante recombinación homóloga residente, debe introducir primero un estado fisiológico especial denominado competencia. La familia de genes RecA/Rad51/DMC1 juega un papel central en la recombinación homóloga durante la transformación bacteriana como lo hace durante la meiosis y la mitosis eucariota. Por ejemplo, la proteína RecA es esencial para la transformación en Bacillus subtilis y Streptococcus pneumoniae,[74] la expresión del gen RecA se induce durante el desarrollo de la competencia para la transformación en estos organismos.
Como parte del proceso de transformación, la proteína RecA interactúa con la introducción de ADN de cadena sencilla (ADNss) para formar nucleofilamentos RecA/ADNss que escanean el cromosoma residente para las regiones de homología y llevan el ADNss entrante a la región correspondiente, donde el intercambio de cadenas y ocurre la recombinación homóloga.[75] Así, el proceso de recombinación homóloga durante la transformación bacteriana tiene similitudes fundamentales a la recombinación homóloga durante la meiosis.
En virus[editar]
La recombinación homóloga se produce en varios grupos de virus. En los virus de ADN, tales como herpesvirus, la recombinación se produce a través de un mecanismo de ruptura y unión como en bacterias y eucariotas.[76] También hay evidencia para la recombinación en algunos virus de ARN, específicamente de sentido positivo de virus ARN de cadena simple como retrovirus, picornavirus, y coronavirus. Existe una controversia sobre si la recombinación homóloga se produce en sentido negativo virus de ARN de cadena simple como la influenza.[77]
En los virus de ARN, la recombinación homóloga puede ser precisa o imprecisa. En el tipo preciso de recombinación RNA-RNA, no hay diferencia entre las dos secuencias de ARN de los padres y la región de ARN de entrecruzamiento resultante. Debido a esto, a menudo es difícil determinar la ubicación de los eventos de cruce entre dos secuencias de ARN recombinantes. En la recombinación homóloga imprecisa de ARN, la región de cruce tiene algunas diferencias con las secuencias de ARN de los padres causado por adición, eliminación u otra modificación de nucleótidos. El nivel de precisión en el cruce es controlado por el contexto de la secuencia de las dos cadenas de recombinación de ARN: secuencias ricas en adenina y uracilo que disminuyen la precisión del cruce.[78][79]
La recombinación homóloga es importante en la facilitación de la evolución viral.[78][80] Por ejemplo, si los genomas de dos virus con diferentes mutaciones desventajosas experimentan recombinación, pueden ser capaces de regenerar un genoma totalmente funcional. Alternativamente, si dos virus similares han infectado a la misma célula huésped, la recombinación homóloga puede permitir que los dos virus intercambien genes y por lo tanto evolucionan variaciones más potentes de sí mismos.[80]
La recombinación homóloga es el mecanismo propuesto por el cual el virus ADN humano herpesvirus-6 se integra en los telómeros humanos.[81]
Cuando dos o más virus, cada uno con un daño genómico letal, infectan la misma célula huésped, los genomas de los virus pueden emparejarse entre sí y se someten a reparación de recombinación homóloga para producir una progenie viable. Este proceso, conocido como reactivación múltiple, se ha estudiado en varios bacteriófagos, incluyendo fago T4.[82] Las enzimas empleadas en la reparación de recombinación en fago T4 son funcionalmente homólogas a las enzimas empleadas en la reparación de recombinación de bacterias y eucariontes.[83] En particular, con lo que se refiere a un gen necesario para la reacción de intercambio de cadena, un paso clave en la reparación de recombinación homóloga, existe una homología funcional de los virus a los seres humanos (es decir, uvsX en fago T4; recA de E. coli y otras bacterias, y rad51 y dmc1 en la levadura y otros eucariontes, incluyendo seres humanos).[84] La reactivación múltiple también ha sido demostrada en numerosos virus patógenos.[85]
Efectos de disfunción[editar]
Sin una recombinación homóloga correcta, los cromosomas se alinean incorrectamente para la primera fase de la división celular en la meiosis. Esto hace que los cromosomas no puedan segregarse adecuadamente en un proceso llamado no disyunción. A su vez, la falta de disyunción puede causar que el esperma y los óvulos tengan muy pocos o demasiados cromosomas. El síndrome de Down, que es causado por una copia extra del cromosoma 21, es una de muchas anormalidades que resultan de un fallo de este tipo de la recombinación homóloga en la meiosis.[67][86]
Las deficiencias en la recombinación homóloga han sido fuertemente ligadas a la formación de cáncer en los seres humanos. Por ejemplo, cada una de las enfermedades relacionadas con el cáncer como el síndrome de Bloom, síndrome de Werner y el síndrome de Rothmund-Thomson son causados por copias que no funcionan correctamente de los genes de la helicasa RecQ implicadas en la regulación de la recombinación homóloga: BLM, WRN y RECQ4, respectivamente.[87] En las células de los pacientes con síndrome de Bloom, que carecen de una copia de la proteína BLM, existe una tasa elevada de recombinación homóloga.[88]
Experimentos en ratones deficientes en BLM han sugerido que la mutación da lugar al cáncer a través de una pérdida de heterocigosidad causada por aumento de la recombinación homóloga.[89] Una pérdida en la heterocigosidad se refiere a la pérdida de una de las dos versiones o de alelos de un gen. Si uno de los alelos perdidos ayuda a suprimir tumores, como el gen de la proteína de retinoblastoma, entonces la pérdida de heterocigosidad puede conducir a cáncer.[90]: 1236
La disminución de las tasas de recombinación homóloga causa la ineficiente reparación del ADN,[90]: 310 que también puede conducir a cáncer.[91] Este es el caso de los genes BRCA1 y BRCA2, dos genes supresores de tumores similares, cuyo mal funcionamiento se ha relacionado con un aumento considerable del riesgo de cáncer de mama y el cáncer de ovario. Las células BRCA1 y BRCA2 que faltan tienen una tasa de disminución de la recombinación homóloga y un aumento de la sensibilidad a la radiación ionizante, lo que sugiere que la disminución de la recombinación homóloga conduce a una mayor susceptibilidad al cáncer.[91] Debido a que la única función conocida de BRCA2 es para ayudar a iniciar la recombinación homóloga, los investigadores han especulado que un conocimiento más detallado de la función de BRCA2 en la recombinación homóloga puede ser la clave para entender las causas del cáncer mama y cáncer de ovario.[91]
Conservación evolutiva[editar]

Si bien las vías mecánicamente pueden variar, la capacidad de los organismos para llevar a cabo la recombinación homóloga es universalmente conservada en todos los dominios de la vida.[92] Basado en la similitud de sus secuencias de aminoácidos, homólogos de unas proteínas se pueden encontrar en varios dominios de la vida que indica que evolucionaron hace mucho tiempo, y desde entonces se separaron de proteínas ancestrales comunes.[92]
Miembros de la familia recombinasa RecA se encuentran en casi todos los organismos con RecA en bacterias, Rad51 y DMC1 en eucariontes, la RadA en arqueas, y UvsX en fago T4.[93]
Proteínas relacionadas de unión de cadena simple que son importantes para la recombinación homóloga, y muchos otros procesos, también se encuentran en todos los dominios de la vida.[94]
Rad54, Mre11, Rad50, y otras proteínas también se encuentran en arqueas y eucariontes.[92][93][95]
La familia RecA recombinasa[editar]
Se cree que las proteínas de la familia recombinasa RecA descienden de una recombinasa ancestral común.[92] La familia recombinasa RecA contiene proteínas RecA de bacterias, las proteínas Rad51 y Dmc1 de eucariotas, y RadA de arqueas, y las proteínas de recombinasa paralog. Estudios que modelan las relaciones evolutivas entre las proteínas Rad51, Dmc1 y RadA indican que son monofiléticas, o que comparten un ancestro común molecular.[92] Dentro de esta familia de proteínas, Rad51 y Dmc1 se agrupan en un clado separado de RadA. Una de las razones para agrupar estas tres proteínas juntas es que todas ellas poseen un motivo hélice-giro-hélice modificado, que ayuda a las proteínas a que se unen al ADN, hacia sus extremos N-terminal.[92] Una antigua duplicación de genes, evento de un gen eucarionte de RecA y la mutación posterior se ha propuesto como un origen probable de los genes RAD51 and DMC1.[92]
Las proteínas generalmente comparten una larga región conservada conocida como el dominio de RecA/Rad51. Dentro del dominio de la proteína hay dos motivos secuenciados, un motivo Walker A y Walker B. Los motivos Walker A y B permiten a los miembros de la familia de proteínas RecA/Rad51 a participar en la unión de ATP y la hidrólisis de ATP.[92][96]
Proteínas específicas de la meiosis[editar]
El descubrimiento de Dmc1 en varias especies de Giardia, uno de los primeros protistas que emergió como un eucarionte, sugiere que la recombinación meiótica homóloga y por lo tanto la meiosis en sí surgieron muy temprano en la evolución eucariota.[97] Además de la investigación en Dmc1, estudios en la proteína Spo11 han proporcionado información sobre los orígenes de la recombinación meiótica.[98] Spo11, una topoisomerasa de tipo II, puede iniciar la recombinación homóloga en la meiosis haciendo rupturas objetivo de dobles cadenas en el ADN.[25] Los árboles filogenéticos basados en la secuencia de genes similares a Spo11 en animales, plantas, hongos, protistas y arqueas han llevado a los científicos a creer que la versión Spo11 actualmente en eucariotas surgió en el último ancestro común de los eucariotas y arqueas.[98]
Aplicaciones tecnológicas[editar]
"Gene targeting"[editar]

Muchos métodos para la introducción de secuencias de ADN en organismos para crear ADN recombinante y organismos modificados genéticamente utilizan el proceso de recombinación homóloga.[99] También llamado "gene targeting", el método es especialmente común en la genética de la levadura y el ratón. El método de "gene targeting" en "ratones knockout" utiliza células madre embrionarias de ratón para entregar material genético artificial (sobre todo de interés terapéutico), que reprime el gen objetivo del ratón por el principio de la recombinación homóloga. El ratón actúa como un modelo de trabajo para entender los efectos de un gen específico de mamífero. En el reconocimiento de su descubrimiento de cómo la recombinación homóloga puede ser utilizada para introducir modificaciones genéticas en ratones a través de células madre embrionarias, Mario Capecchi, Martin Evans y Oliver Smithies fueron galardonados con el Premio Nobel 2007 de Fisiología o Medicina.[100]
Los avances en las tecnologías de "gene targeting" que secuestran los mecanismos de recombinación homóloga de células están llevando al desarrollo de una nueva ola de modelos más precisos, modelos isogénicos humanos de las enfermedades. Se cree que estos modelos celulares humanos son pensados para reflejar con mayor exactitud la genética de enfermedades humanas que el modelo de ratón predecesor. Esto es en gran parte debido a que las mutaciones de interés se introducen en los genes endógenos, tal como se presentan en los pacientes reales, y porque se basan en los genomas humanos en lugar de los genomas de ratas. Por otra parte, ciertas tecnologías permiten el "knock-in" de una mutación particular en lugar de solo un "knock-out" de genes asociados con las tecnologías más antiguas de "gene targeting".
Ingeniería de proteínas[editar]
La ingeniería de proteínas con la recombinación homóloga desarrolla proteínas quiméricas mediante el cambio de fragmentos entre dos proteínas parentales. Estas técnicas explotan el hecho de que la recombinación puede introducir un alto grado de diversidad de secuencia preservando al mismo tiempo la capacidad de una proteína para doblar a su estructura terciaria, o forma tridimensional.[101] Esto está en contraste con otras técnicas de ingeniería de proteínas, como el punto de mutagénesis aleatoria, en la que la probabilidad de mantener la función de proteínas disminuye exponencialmente con el aumento de las sustituciones de aminoácidos.[102] Las quimeras producidas por técnicas de recombinación son capaces de mantener su capacidad de doblaje porque sus fragmentos parentales intercambiados se conservan estructuralmente y evolutivamente. Estos "bloques de construcción" recombinables conservan interacciones estructuralmente importantes como puntos de contacto físico entre los diferentes aminoácidos en la estructura de la proteína. Métodos computacionales como SCHEMA y el análisis estadístico de acoplamiento se pueden utilizar para identificar subunidades estructurales adecuadas para la recombinación.[103][104][105]
Las técnicas que se basan en la recombinación homóloga se han utilizado para diseñar nuevas proteínas.[103] En un estudio publicado en el 2007, los investigadores fueron capaces de crear quimeras de dos enzimas implicadas en la biosíntesis de isoprenoides, una diversa clase de compuestos incluyendo hormonas, pigmentos visuales y ciertas feromonas. Las proteínas quiméricas adquirieron la capacidad de catalizar una reacción esencial en la biosíntesis de isoprenoides, una de las vías más diversas de biosíntesis que se encuentran en la naturaleza, que estuvo ausente en las proteínas de origen parental.[106]
La ingeniería de proteínas a través de la recombinación también ha producido enzimas quiméricas con una nueva función en los miembros de un grupo de proteínas conocidas como la familia del citocromo P450,[107] que en los humanos está implicada en la desintoxicación de compuestos extraños como las drogas, aditivos y conservadores de alimentos.[22]
Terapia de cáncer[editar]
Las células cancerosas con mutaciones BRCA tienen deficiencias en la recombinación homóloga, y drogas para explotar esas deficiencias se han desarrollado y utilizado con éxito en ensayos clínicos.[108][109] Olaparib, un inhibidor de PARP1, encoge o detiene el crecimiento de tumores de mama, de ovario y los cánceres de próstata causados por mutaciones en los genes BRCA1 o BRCA2, que son necesarios para RH. Cuando los genes BRCA1 o BRCA2 están ausente, otros tipos de mecanismos de reparación del ADN deben compensar la carencia de recombinación homóloga, como la reparación por escisión de bases (BER) para las horquillas de replicación atascadas o de extremos no homólogos (NHEJ) para rupturas de doble cadena.[108] Al inhibir BER en una celda de RH-deficiente, Olaparib aplica el concepto de letalidad sintética para dirigirse específicamente a las células cancerosas. Mientras que los inhibidores PARP1 representan un enfoque novedoso para el tratamiento del cáncer, los investigadores han advertido que, pueden resultar insuficientes para el tratamiento de cánceres metastásicos en etapa tardía.[108] Las células cancerosas pueden volverse resistentes a un inhibidor de PARP1 si se someten a deleciones de mutaciones en BRCA2, lo que rompe la letalidad sintética de drogas mediante la restauración de la capacidad de las células cancerosas para reparar el ADN por RH.[110]
Referencias[editar]
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2002). «Chapter 5: DNA Replication, Repair, and Recombination». Molecular Biology of the Cell (4th edición). New York: Garland Science. p. 845. ISBN 0-8153-3218-1. OCLC 145080076.
- ↑ Capecchi, M. R. (16 de junio de 1989). «Altering the genome by homologous recombination». Science (New York, N.Y.) 244 (4910): 1288-1292. ISSN 0036-8075. PMID 2660260.
- ↑ Smithies, O.; Gregg, R. G.; Boggs, S. S.; Koralewski, M. A.; Kucherlapati, R. S. (19 de septiembre de 1985). «Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination». Nature 317 (6034): 230-234. ISSN 0028-0836. PMID 2995814.
- ↑ Orr-Weaver, T. L.; Szostak, J. W.; Rothstein, R. J. (1 de octubre de 1981). «Yeast transformation: a model system for the study of recombination». Proceedings of the National Academy of Sciences of the United States of America 78 (10): 6354-6358. ISSN 0027-8424. PMC 349037. PMID 6273866.
- ↑ Orr-Weaver, T. L.; Szostak, J. W. (1 de julio de 1983). «Yeast recombination: the association between double-strand gap repair and crossing-over». Proceedings of the National Academy of Sciences of the United States of America 80 (14): 4417-4421. ISSN 0027-8424. PMC 384049. PMID 6308623.
- ↑ Szostak, J. W.; Orr-Weaver, T. L.; Rothstein, R. J.; Stahl, F. W. (1 de mayo de 1983). «The double-strand-break repair model for recombination». Cell 33 (1): 25-35. ISSN 0092-8674. PMID 6380756.
- ↑ Resnick, M. A. (1 de junio de 1976). «The repair of double-strand breaks in DNA; a model involving recombination». Journal of Theoretical Biology 59 (1): 97-106. ISSN 0022-5193. PMID 940351.
- ↑ Jasin, Maria; Rothstein, Rodney (1 de noviembre de 2013). «Repair of Strand Breaks by Homologous Recombination». Cold Spring Harbor Perspectives in Biology 5 (11). ISSN 1943-0264. PMC 3809576. PMID 24097900. doi:10.1101/cshperspect.a012740.
- ↑ Bateson P (Aug 2002). «William Bateson: a biologist ahead of his time». Journal of Genetics 81 (2): 49-58. PMID 12532036. doi:10.1007/BF02715900.
- ↑ «Reginald Crundall Punnett». NAHSTE, University of Edinburgh. Consultado el 3 de julio de 2010.
- ↑ Lobo, I; Shaw, K (2008). «Thomas Hunt Morgan, genetic recombination, and gene mapping». Nature Education 1 (1).
- ↑ a b Coe E; Kass LB (May 2005). «Proof of physical exchange of genes on the chromosomes». Proceedings of the National Academy of Sciences 102 (19): 6641-6. PMC 1100733. PMID 15867161. doi:10.1073/pnas.0407340102.
- ↑ Creighton HB; McClintock B (Aug 1931). «A Correlation of Cytological and Genetical Crossing-Over in Zea Mays». Proceedings of the National Academy of Sciences 17 (8): 492-7. PMC 1076098. PMID 16587654. doi:10.1073/pnas.17.8.492.
- ↑ Stern, C (1931). «Zytologisch-genetische untersuchungen alsbeweise fur die Morgansche theorie des faktoraustauschs». Biol. Zentbl. 51: 547-587.
- ↑ «The development of bacterial genetics». US National Library of Medicine. Consultado el 3 de julio de 2010.
- ↑ «The Nobel Prize in Physiology or Medicine 1958». Nobelprize.org. Consultado el 3 de julio de 2010.
- ↑ a b c Haber JE; Ira G; Malkova A; Sugawara N (Jan 2004). «Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday's model». Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 359 (1441): 79-86. PMC 1693306. PMID 15065659. doi:10.1098/rstb.2003.1367.
- ↑ Szostak JW; Orr-Weaver TL; Rothstein RJ; Stahl FW (May 1983). «The double-strand-break repair model for recombination». Cell 33 (1): 25-35. PMID 6380756. doi:10.1016/0092-8674(83)90331-8.
- ↑ Lodish H; Berk A; Zipursky SL; Matsudaira P; Baltimore D; Darnell J (2000). «12.5: Recombination between Homologous DNA Sites: Double-Strand Breaks in DNA Initiate Recombination». Molecular Cell Biology (4th edición). W. H. Freeman and Company. ISBN 0-7167-3136-3.
- ↑ Griffiths, AJF (1999). «8: Chromosome Mutations: Chromosomal Rearrangements». Modern Genetic Analysis. W. H. Freeman and Company. ISBN 0-7167-3118-5.
- ↑ Khanna KK; Jackson SP (Mar 2001). «DNA double-strand breaks: signaling, repair and the cancer connection». Nature Genetics 27 (3): 247-54. PMID 11242102. doi:10.1038/85798.
- ↑ a b c Nelson, DL; Cox, MM (2005). Principles of Biochemistry (4th edición). Freeman. pp. 980–981. ISBN 978-0-7167-4339-2.
- ↑ Marcon E; Moens PB (Aug 2005). «The evolution of meiosis: recruitment and modification of somatic DNA-repair proteins». BioEssays 27 (8): 795-808. PMID 16015600. doi:10.1002/bies.20264.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2008). Molecular Biology of the Cell (5th edición). Garland Science. p. 305. ISBN 978-0-8153-4105-5.
- ↑ a b Keeney S; Giroux CN; Kleckner N (Feb 1997). «Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family». Cell 88 (3): 375-84. PMID 9039264. doi:10.1016/S0092-8674(00)81876-0.
- ↑ Longhese MP; Bonetti D; Guerini I; Manfrini N; Clerici M (Sep 2009). «DNA double-strand breaks in meiosis: checking their formation, processing and repair». DNA Repair 8 (9): 1127-38. PMID 19464965. doi:10.1016/j.dnarep.2009.04.005.
- ↑ Cahill LP; Mariana JC; Mauléon P (Jan 1979). «Total follicular populations in ewes of high and low ovulation rates». Journal of Reproduction and Fertility 55 (1): 27-36. PMC 423159. doi:10.1371/journal.pbio.0020192.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2008). Molecular Biology of the Cell (5th edición). Garland Science. p. 303. ISBN 978-0-8153-4105-5.
- ↑ a b Shrivastav M; De Haro LP; Nickoloff JA (Jan 2008). «Regulation of DNA double-strand break repair pathway choice». Cell Research 18 (1): 134-47. PMID 18157161. doi:10.1038/cr.2007.111.
- ↑ a b Mimitou EP; Symington LS (May 2009). «Nucleases and helicases take center stage in homologous recombination». Trends in Biochemical Sciences 34 (5): 264-72. PMID 19375328. doi:10.1016/j.tibs.2009.01.010.
- ↑ Huertas P; Cortés-Ledesma F, F; Sartori AA, AA; Aguilera A; Jackson SP (Oct 2008). «CDK targets Sae2 to control DNA-end resection and homologous recombination». Nature 455 (7213): 689-92. PMC 2635538. PMID 18716619. doi:10.1038/nature07215.
- ↑ a b c d Sung P; Klein H, H (Oct 2006). «Mechanism of homologous recombination: mediators and helicases take on regulatory functions». Nature Reviews. Molecular Cell Biology 7 (10): 739-50. PMID 16926856. doi:10.1038/nrm2008.
- ↑ Wold MS (1997). «Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism». Annual Review of Biochemistry 66: 61-92. PMID 9242902. Texto « doi 10.1146/annurev.biochem.66.1.61 » ignorado (ayuda)
- ↑ McMahill MS; Sham CW; Bishop DK (Nov 2007). «Synthesis-dependent strand annealing in meiosis». PLoS Biology 5 (11): e299. PMC 2062477. PMID 17988174. doi:10.1371/journal.pbio.0050299.
- ↑ Bärtsch S; Kang LE; Symington LS (Feb 2000). «RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates». Molecular and Cellular Biology 20 (4): 1194-205. PMC 85244. PMID 10648605. doi:10.1128/MCB.20.4.1194-1205.2000.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2008). Molecular Biology of the Cell (5th edición). Garland Science. pp. 312-313. ISBN 978-0-8153-4105-5.
- ↑ a b c Helleday T; Lo J, J; van Gent DC, DC; Engelward BP (Jul 2007). «DNA double-strand break repair: from mechanistic understanding to cancer treatment». DNA Repair 6 (7): 923-35. PMID 17363343. doi:10.1016/j.dnarep.2007.02.006.
- ↑ a b Andersen SL; Sekelsky J (Dec 2010). «Meiotic versus mitotic recombination: two different routes for double-strand break repair: the different functions of meiotic versus mitotic DSB repair are reflected in different pathway usage and different outcomes». BioEssays 32 (12): 1058-66. PMC 3090628. PMID 20967781. doi:10.1002/bies.201000087.
- ↑ Allers T; Lichten M (Jul 2001). «Differential timing and control of noncrossover and crossover recombination during meiosis». Cell 106 (1): 47-57. PMID 11461701. doi:10.1016/s0092-8674(01)00416-0.
- ↑ Haber lab. «Single-strand annealing». "Brandeis University". Consultado el 3 de julio de 2010.
- ↑ a b c Lyndaker AM; Alani E, E (Mar 2009). «A tale of tails: insights into the coordination of 3' end processing during homologous recombination». BioEssays 31 (3): 315-21. PMC 2958051. PMID 19260026. doi:10.1002/bies.200800195.
- ↑ Mimitou EP; Symington LS (Sep 2009). «DNA end resection: many nucleases make light work». DNA Repair 8 (9): 983-95. PMC 2760233. PMID 19473888. doi:10.1016/j.dnarep.2009.04.017.
- ↑ Pâques F; Haber JE (Jun 1999). «Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae». Microbiology and Molecular Biology Reviews 63 (2): 349-404. PMC 98970. PMID 10357855.
- ↑ a b McEachern MJ; Haber JE (2006). «Break-induced replication and recombinational telomere elongation in yeast». Annual Review of Biochemistry 75: 111-35. PMID 16756487. doi:10.1146/annurev.biochem.74.082803.133234.
- ↑ Morrish TA; Greider CW (Jan 2009). «Short telomeres initiate telomere recombination in primary and tumor cells». En Haber, James E., ed. PLoS Genetics 5 (1): e1000357. PMC 2627939. PMID 19180191. doi:10.1371/journal.pgen.1000357.
- ↑ Muntoni A; Reddel RR (Oct 2005). «The first molecular details of ALT in human tumor cells». Human Molecular Genetics. 14 Spec No. 2 (Review Issue 2): R191-6. PMID 16244317. doi:10.1093/hmg/ddi266.
- ↑ PDB 3cmt
- Chen Z; ang H; Pavletich NP (May 2008). «Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures». Nature 453 (7194): 489-4. PMID 18497818. doi:10.1038/nature06971.
- ↑ Kowalczykowski SC; Dixon DA, DA; Eggleston AK, AK; Lauder SD; Rehrauer WM (Sep 1994). «Biochemistry of homologous recombination in Escherichia coli». Microbiological Reviews 58 (3): 401-65. PMC 372975. PMID 7968921.
- ↑ Rocha EP; Cornet E, E; Michel B, B (Aug 2005). «Comparative and evolutionary analysis of the bacterial homologous recombination systems». PLoS Genetics 1 (2): e15. PMC 1193525. PMID 16132081. doi:10.1371/journal.pgen.0010015.
- ↑ Amundsen SK; Taylor AF, A. F.; Reddy M, M.; Smith GR (Dec 2007). «Intersubunit signaling in RecBCD enzyme, a complex protein machine regulated by Chi hot spots». Genes & Development 21 (24): 3296-307. PMC 2113030. PMID 18079176. doi:10.1101/gad.1605807.
- ↑ Cromie GA (Aug 2009). «Phylogenetic ubiquity and shuffling of the bacterial RecBCD and AddAB recombination complexes». Journal of Bacteriology 191 (16): 5076-84. PMC 2725590. PMID 19542287. doi:10.1128/JB.00254-09.
- ↑ a b Smith GR (Jun 2012). «How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist's view». Microbiology and Molecular Biology Reviews 76 (2): 217-28. PMID 22688812. doi:10.1128/MMBR.05026-11.
- ↑ a b c d Dillingham MS; Kowalczykowski SC (Dec 2008). «RecBCD enzyme and the repair of double-stranded DNA breaks». Microbiology and Molecular Biology Reviews 72 (4): 642-71, Table of Contents. PMC 2593567. PMID 19052323. doi:10.1128/MMBR.00020-08.
- ↑ Michel B; Boubakri H; Baharoglu Z; LeMasson M; Lestini R (Jul 2007). «Recombination proteins and rescue of arrested replication forks». DNA Repair 6 (7): 967-80. PMID 17395553. doi:10.1016/j.dnarep.2007.02.016.
- ↑ Amundsen SK; Taylor AF, AF; Reddy M, M; Smith GR (Dec 2007). «Intersubunit signaling in RecBCD enzyme, a complex protein machine regulated by Chi hot spots». Genes & Development 21 (24): 3296-307. PMC 2113030. PMID 18079176. doi:10.1101/gad.1605807.
- ↑ Spies M; Bianco PR, PR; Dillingham MS, MS; Handa N; Baskin RJ; Kowalczykowski SC (Sep 2003). «A molecular throttle: the recombination hotspot chi controls DNA translocation by the RecBCD helicase». Cell 114 (5): 647-54. PMID 13678587. doi:10.1016/S0092-8674(03)00681-0.
- ↑ Taylor AF; Smith GR, GR (Jun 2003). «RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity». Nature 423 (6942): 889-93. PMID 12815437. doi:10.1038/nature01674.
- ↑ Spies M; Amitani I, I; Baskin RJ, RJ; Kowalczykowski SC (Nov 2007). «RecBCD enzyme switches lead motor subunits in response to chi recognition». Cell 131 (4): 694-705. PMC 2151923. PMID 18022364. doi:10.1016/j.cell.2007.09.023.
- ↑ Savir Y; Tlusty T (Nov 2010). «RecA-mediated homology search as a nearly optimal signal detection system». Molecular Cell 40 (3): 388-96. PMID 21070965. doi:10.1016/j.molcel.2010.10.020. Archivado desde el original el 7 de octubre de 2012. Consultado el 16 de junio de 2016.
- ↑ Rambo RP; Williams GJ; Tainer JA (Nov 2010). «Achieving fidelity in homologous recombination despite extreme complexity: informed decisions by molecular profiling». Molecular Cell 40 (3): 347-8. PMC 3003302. PMID 21070960. doi:10.1016/j.molcel.2010.10.032. Archivado desde el original el 7 de octubre de 2012. Consultado el 16 de junio de 2016.
- ↑ De Vlaminck I; van Loenhout MT; Zweifel L; den Blanken J; Hooning K; Hage S; Kerssemakers J; Dekker C (Jun 2012). «Mechanism of homology recognition in DNA recombination from dual-molecule experiments». Molecular Cell 46 (5): 616-24. PMID 22560720. doi:10.1016/j.molcel.2012.03.029.
- ↑ Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2008). Molecular Biology of the Cell (5th edición). Garland Science. p. 307. ISBN 978-0-8153-4105-5.
- ↑ Morimatsu K; Kowalczykowski SC (May 2003). «RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair». Molecular Cell 11 (5): 1337-47. PMID 12769856. doi:10.1016/S1097-2765(03)00188-6.
- ↑ Hiom K (Jul 2009). «DNA repair: common approaches to fixing double-strand breaks». Current Biology 19 (13): R523-5. PMID 19602417. doi:10.1016/j.cub.2009.06.009.
- ↑ Handa N; Morimatsu K, K; Lovett ST, ST; Kowalczykowski SC (May 2009). «Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli». Genes & Development 23 (10): 1234-45. PMC 2685532. PMID 19451222. doi:10.1101/gad.1780709.
- ↑ a b West SC (Jun 2003). «Molecular views of recombination proteins and their control». Nature Reviews. Molecular Cell Biology 4 (6): 435-45. PMID 12778123. doi:10.1038/nrm1127.
- ↑ a b c d Watson, James D.; Baker, Tania A.; Bell, Stephen P.; Gann, Alexander; Levine, Michael; Losick, Richard (2003). Molecular Biology of the Gene (5th edición). Pearson/Benjamin Cummings. pp. 259–291. ISBN 978-0-8053-4635-0.
- ↑ Gumbiner-Russo LM; Rosenberg SM (28 de noviembre de 2007). «Physical analyses of E. coli heteroduplex recombination products in vivo: on the prevalence of 5' and 3' patches». En Sandler, Steve, ed. PloS One 2 (11): e1242. PMC 2082072. PMID 18043749. doi:10.1371/journal.pone.0001242.
- ↑ Thomas CM; Nielsen KM (Sep 2005). «Mechanisms of, and barriers to, horizontal gene transfer between bacteria». Nature Reviews. Microbiology 3 (9): 711-21. PMID 16138099. doi:10.1038/nrmicro1234. Archivado desde el original el 1 de junio de 2010.
- ↑ Vulić M; Dionisio F, F; Taddei F, F; Radman M (Sep 1997). «Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria». Proceedings of the National Academy of Sciences 94 (18): 9763-7. PMC 23264. PMID 9275198. doi:10.1073/pnas.94.18.9763.
- ↑ Majewski J; Cohan FM (Jan 1998). «The effect of mismatch repair and heteroduplex formation on sexual isolation in Bacillus». Genetics 148 (1): 13-8. PMC 1459767. PMID 9475717.
- ↑ Majewski J; Zawadzki P, P; Pickerill P, P; Cohan FM; Dowson CG (Feb 2000). «Barriers to genetic exchange between bacterial species: Streptococcus pneumoniae transformation». Journal of Bacteriology 182 (4): 1016-23. PMC 94378. PMID 10648528. doi:10.1128/JB.182.4.1016-1023.2000.
- ↑ Chen I; Dubnau D (Mar 2004). «DNA uptake during bacterial transformation». Nature Reviews. Microbiology 2 (3): 241-9. PMID 15083159. doi:10.1038/nrmicro844.
- ↑ Claverys JP; Martin B; Polard P (May 2009). «The genetic transformation machinery: composition, localization, and mechanism». FEMS Microbiology Reviews 33 (3): 643-56. PMID 19228200. doi:10.1111/j.1574-6976.2009.00164.x.
- ↑ Kidane D; Graumann PL (Jul 2005). «Intracellular protein and DNA dynamics in competent Bacillus subtilis cells». Cell 122 (1): 73-84. PMID 16009134. doi:10.1016/j.cell.2005.04.036.
- ↑ Fleischmann Jr, WR (1996). «43». Medical Microbiology (4th edición). University of Texas Medical Branch at Galveston. ISBN 0-9631172-1-1.
- ↑ Boni MF; de Jong MD; van Doorn HR; Holmes EC (3 de mayo de 2010). «Guidelines for identifying homologous recombination events in influenza A virus». En Martin, Darren P., ed. PloS One 5 (5): e10434. PMC 2862710. PMID 20454662. doi:10.1371/journal.pone.0010434.
- ↑ a b Nagy PD; Bujarski JJ (Jan 1996). «Homologous RNA recombination in brome mosaic virus: AU-rich sequences decrease the accuracy of crossovers». Journal of Virology 70 (1): 415-26. PMC 189831. PMID 8523555.
- ↑ Chetverin AB (Oct 1999). «The puzzle of RNA recombination». FEBS Letters 460 (1): 1-5. PMID 10571050. doi:10.1016/S0014-5793(99)01282-X.
- ↑ a b Roossinck MJ (September 1997). «Mechanisms of plant virus evolution». Annual Review of Phytopathology 35: 191-209. PMID 15012521. doi:10.1146/annurev.phyto.35.1.191.
- ↑ Arbuckle JH; Medveczky PG, Peter G. (Aug 2011). «The molecular biology of human herpesvirus-6 latency and telomere integration». Microbes and Infection / Institut Pasteur 13 (8-9): 731-41. PMC 3130849. PMID 21458587. doi:10.1016/j.micinf.2011.03.006.
- ↑ Bernstein C (Mar 1981). «Deoxyribonucleic acid repair in bacteriophage». Microbiological Reviews 45 (1): 72-98. PMC 281499. PMID 6261109.
- ↑ Bernstein C, Bernstein H (2001). DNA repair in bacteriophage. In: Nickoloff JA, Hoekstra MF (Eds.) DNA Damage and Repair, Vol.3. Advances from Phage to Humans. Humana Press, Totowa, NJ, pp. 1–19. ISBN 978-0896038035
- ↑ Story RM; Bishop DK; Kleckner N; Steitz TA (Mar 1993). «Structural relationship of bacterial RecA proteins to recombination proteins from bacteriophage T4 and yeast». Science 259 (5103): 1892-6. PMID 8456313. doi:10.1126/science.8456313.
- ↑ Michod RE; Bernstein H; Nedelcu AM (May 2008). «Adaptive value of sex in microbial pathogens». Infection, Genetics and Evolution 8 (3): 267-85. PMID 18295550. doi:10.1016/j.meegid.2008.01.002.http://www.hummingbirds.arizona.edu/Faculty/Michod/Downloads/IGE%20review%20sex.pdf
- ↑ Lamb NE; Yu K, Shaffer J, K; Feingold E, J; Sherman SL (Jan 2005). «Association between maternal age and meiotic recombination for trisomy 21». American Journal of Human Genetics 76 (1): 91-9. PMC 1196437. PMID 15551222. doi:10.1086/427266.
- ↑ Cold Spring Harbor Laboratory (2007). «Human RecQ Helicases, Homologous Recombination And Genomic Instability». ScienceDaily. Consultado el 3 de julio de 2010.
- ↑ Modesti M; Kanaar R (2001). «Homologous recombination: from model organisms to human disease». Genome Biology 2 (5): REVIEWS1014. PMC 138934. PMID 11387040. doi:10.1186/gb-2001-2-5-reviews1014.
- ↑ Luo G; Santoro IM; McDaniel LD; Nishijima I; Mills M; Youssoufian H; Vogel H; Schultz RA et al. (Dec 2000). «Cancer predisposition caused by elevated mitotic recombination in Bloom mice». Nature Genetics 26 (4): 424-9. PMID 11101838. doi:10.1038/82548.
- ↑ a b Alberts, Bruce; Johnson, Alexander; Lewis, Julian; Raff, Martin; Roberts, Keith; Walter, Peter (2007). Molecular Biology of the Cell (5th edición). Garland Science. ISBN 978-0-8153-4110-9.
- ↑ a b c Powell SN; Kachnic LA (Sep 2003). «Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation». Oncogene 22 (37): 5784-91. PMID 12947386. doi:10.1038/sj.onc.1206678.
- ↑ a b c d e f g h Lin Z; Kong H, H; Nei M, M; Ma H (Jul 2006). «Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer». Proceedings of the National Academy of Sciences 103 (27): 10328-33. PMC 1502457. PMID 16798872. doi:10.1073/pnas.0604232103.
- ↑ a b Haseltine CA; Kowalczykowski SC (May 2009). «An archaeal Rad54 protein remodels DNA and stimulates DNA strand exchange by RadA». Nucleic Acids Research 37 (8): 2757-70. PMC 2677860. PMID 19282450. doi:10.1093/nar/gkp068.
- ↑ Rolfsmeier ML; Haseltine CA (Mar 2010). «The single-stranded DNA binding protein of Sulfolobus solfataricus acts in the presynaptic step of homologous recombination». Journal of Molecular Biology 397 (1): 31-45. PMID 20080104. doi:10.1016/j.jmb.2010.01.004.
- ↑ Huang Q; Liu L; Liu J; Ni J; She Q; Shen Y (2015). «Efficient 5'-3' DNA end resection by HerA and NurA is essential for cell viability in the crenarchaeon Sulfolobus islandicus». BMC Molecular Biology 16: 2. PMC 4351679. PMID 25880130. doi:10.1186/s12867-015-0030-z.
- ↑ Jain SK; Cox MM; Inman RB (Aug 1994). «On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. III. Unidirectional branch migration and extensive hybrid DNA formation». The Journal of Biological Chemistry 269 (32): 20653-61. PMID 8051165.
- ↑ Ramesh MA; Malik SB; Logsdon JM (Jan 2005). «A phylogenomic inventory of meiotic genes; evidence for sex in Giardia and an early eukaryotic origin of meiosis». Current Biology 15 (2): 185-91. PMID 15668177. doi:10.1016/j.cub.2005.01.003.
- ↑ a b Malik SB; Ramesh MA; Hulstrand AM; Logsdon JM (Dec 2007). «Protist homologs of the meiotic Spo11 gene and topoisomerase VI reveal an evolutionary history of gene duplication and lineage-specific loss». Molecular Biology and Evolution 24 (12): 2827-41. PMID 17921483. doi:10.1093/molbev/msm217.
- ↑ Lodish H; Berk A; Zipursky SL; Matsudaira P; Baltimore D; Darnell J (2000). «Chapter 8.5: Gene Replacement and Transgenic Animals: DNA Is Transferred into Eukaryotic Cells in Various Ways». Molecular Cell Biology (4th edición). W. H. Freeman and Company. ISBN 0-7167-3136-3.
- ↑ «The Nobel Prize in Physiology or Medicine 2007». The Nobel Foundation. Consultado el 15 de diciembre de 2008.
- ↑ Drummond DA; Silberg JJ, JJ; Meyer MM, MM; Wilke CO; Arnold FH (Apr 2005). «On the conservative nature of intragenic recombination». Proceedings of the National Academy of Sciences 102 (15): 5380-5. PMC 556249. PMID 15809422. doi:10.1073/pnas.0500729102.
- ↑ Bloom JD; Silberg JJ, JJ; Wilke CO, CO; Drummond DA; Adami C; Arnold FH (Jan 2005). «Thermodynamic prediction of protein neutrality». Proceedings of the National Academy of Sciences 102 (3): 606-11. PMC 545518. PMID 15644440. doi:10.1073/pnas.0406744102.
- ↑ a b Carbone MN; Arnold FH (Aug 2007). «Engineering by homologous recombination: exploring sequence and function within a conserved fold». Current Opinion in Structural Biology 17 (4): 454-9. PMID 17884462. doi:10.1016/j.sbi.2007.08.005.
- ↑ Otey CR; Landwehr M, M; Endelman JB, JB; Hiraga K; Bloom JD; Arnold FH (May 2006). «Structure-guided recombination creates an artificial family of cytochromes P450». PLoS Biology 4 (5): e112. PMC 1431580. PMID 16594730. doi:10.1371/journal.pbio.0040112.
- ↑ Socolich M; Lockless SW, SW; Russ WP, WP; Lee H; Gardner KH; Ranganathan R (Sep 2005). «Evolutionary information for specifying a protein fold». Nature 437 (7058): 512-8. PMID 16177782. doi:10.1038/nature03991.
- ↑ Thulasiram HV; Erickson HK; Poulter CD (Apr 2007). «Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis». Science 316 (5821): 73-6. PMID 17412950. doi:10.1126/science.1137786.
- ↑ Landwehr M; Carbone M, M; Otey CR, CR; Li Y; Arnold FH (Mar 2007). «Diversification of catalytic function in a synthetic family of chimeric cytochrome p450s». Chemistry & Biology 14 (3): 269-78. PMC 1991292. PMID 17379142. doi:10.1016/j.chembiol.2007.01.009.
- ↑ a b c Iglehart JD; Silver DP (Jul 2009). «Synthetic lethality--a new direction in cancer-drug development». The New England Journal of Medicine 361 (2): 189-91. PMID 19553640. doi:10.1056/NEJMe0903044.
- ↑ Fong PC; Boss DS; Yap TA; Tutt A; Wu P; Mergui-Roelvink M; Mortimer P; Swaisland H; Lau A; O'Connor MJ; Ashworth A; Carmichael J; Kaye SB; Schellens JH; de Bono JS (Jul 2009). «Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers». The New England Journal of Medicine 361 (2): 123-34. PMID 19553641. doi:10.1056/NEJMoa0900212.
- ↑ Edwards SL; Brough R, R; Lord CJ, CJ; Natrajan R; Vatcheva R; Levine DA; Boyd J; Reis-Filho JS et al. (Feb 2008). «Resistance to therapy caused by intragenic deletion in BRCA2». Nature 451 (7182): 1111-5. PMID 18264088. doi:10.1038/nature06548.
Enlaces externos[editar]
 Wikimedia Commons alberga una categoría multimedia sobre Recombinación homóloga.
Wikimedia Commons alberga una categoría multimedia sobre Recombinación homóloga.- Animations – homologous recombination: Animaciones que muestran muchos modelos de recombinación homóloga.
- Homologous recombination: Tempy & Trun: Animación de la vía bacteriana RecBCD de recombinación homóloga.
| Control de autoridades |
|
|---|
 Datos: Q22280388
Datos: Q22280388- Multimedia: Homologous recombination / Q22280388